We strive to develop innovative therapies by strategically focusing on genetically defined diseases. Our clinical pipeline is focused on bezuclastinib, a highly selective and potent KIT inhibitor with the potential to provide real solutions for patients. Our research pipeline leverages our world-class research and development team to advance our preclinical assets.
Clinical Programs

Bezuclastinib, a tyrosine kinase inhibitor (TKI) selectively targeting KIT activation loop mutations in systemic mastocytosis (SM)
APEX is a registration-directed, global, open-label trial in patients with advanced systemic mastocytosis (AdvSM).
Learn More About:
Bezuclastinib, a tyrosine kinase inhibitor (TKI) selectively targeting KIT activation loop mutations in systemic mastocytosis (SM)
SUMMIT is a registration-directed, randomized, double-blind, placebo-controlled, global, multicenter, clinical trial of bezuclastinib in patients with nonadvanced systemic mastocytosis (NonAdvSM).
Learn More About:
Bezuclastinib, a tyrosine kinase inhibitor selectively targeting KIT activation loop mutations in GIST
PEAK is a global, blinded, randomized Phase 3 clinical trial studying the combination of bezuclastinib and sunitinib versus sunitinib alone in patients with imatinib-resistant gastrointestinal stromal tumors (GIST).
Learn More About:
Disease Background
- FGFRs regulate several cellular processes such as proliferation, migration, survival and angiogenesis by binding to FGFs2
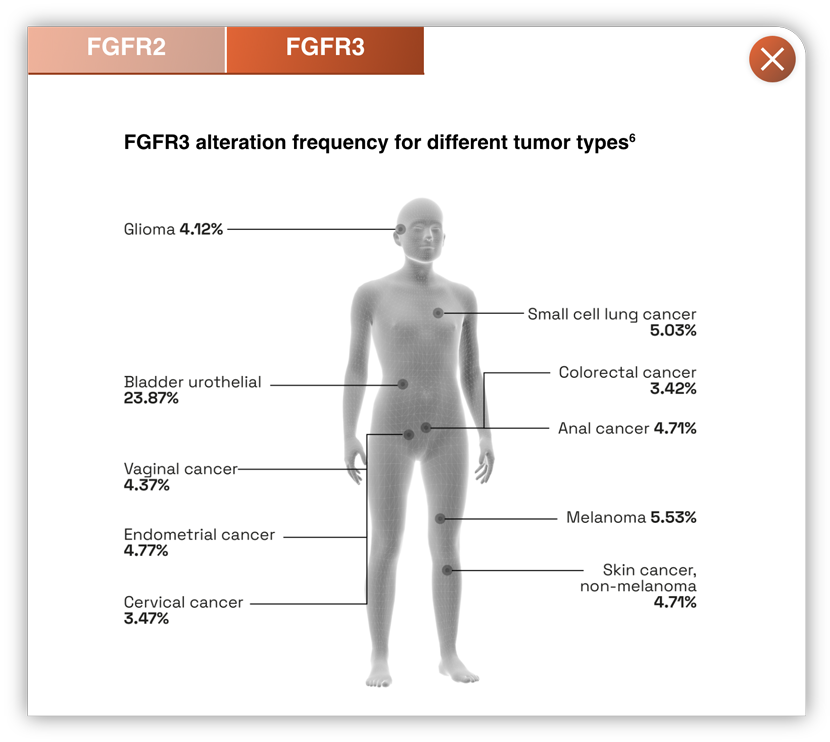
- FGFR aberrations cause abnormal signalling and are responsible for driving a wide variety of cancer types2
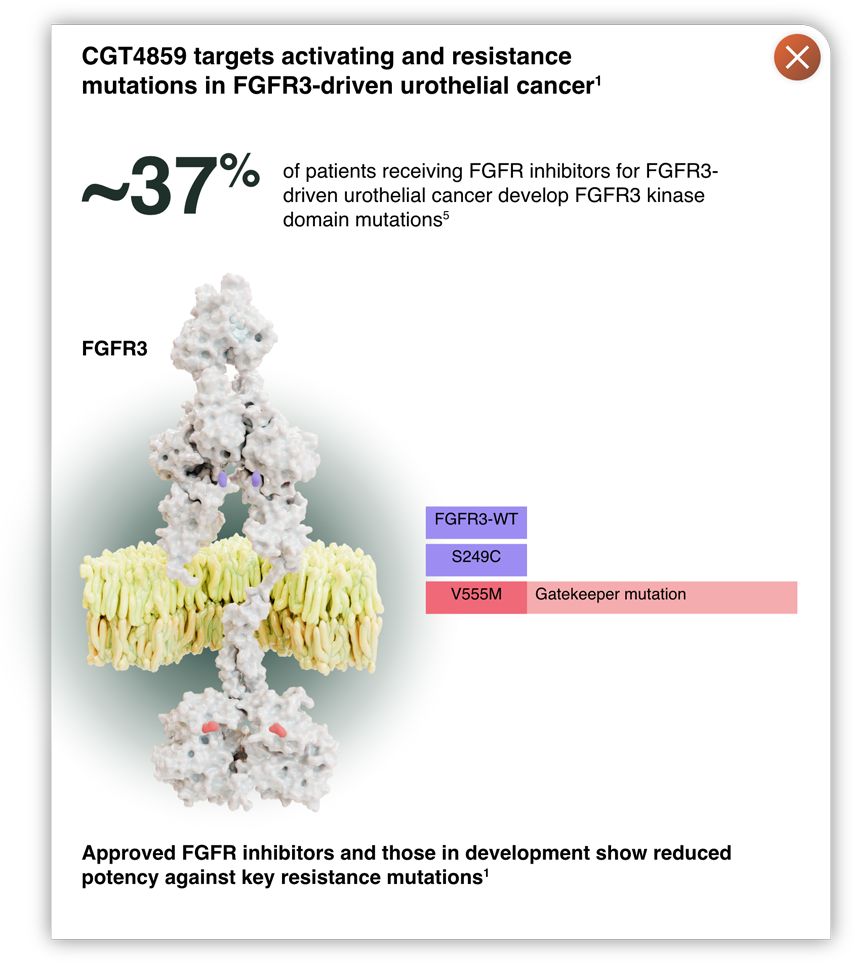
- Inhibition of FGFR3 mutations in urothelial cancer, and FGFR2 fusions in cholangiocarcinoma, has led to improved clinical outcomes1
THE UNMET NEED IN FGFR INHIBITION
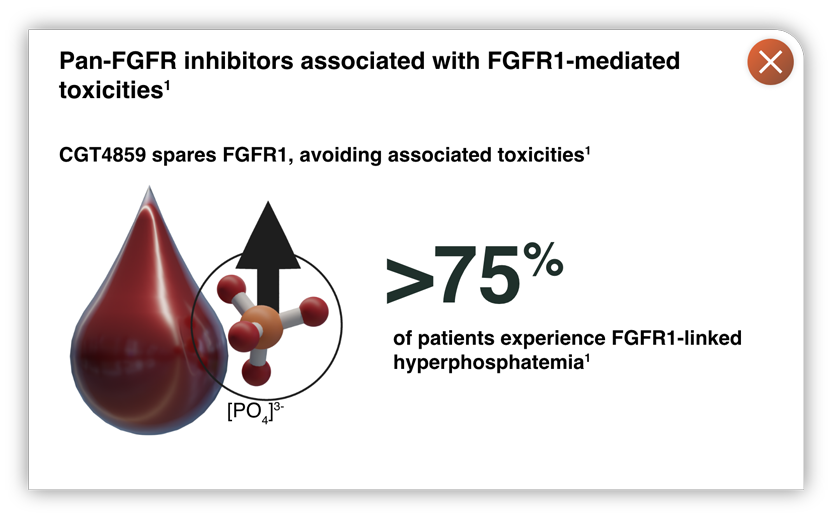
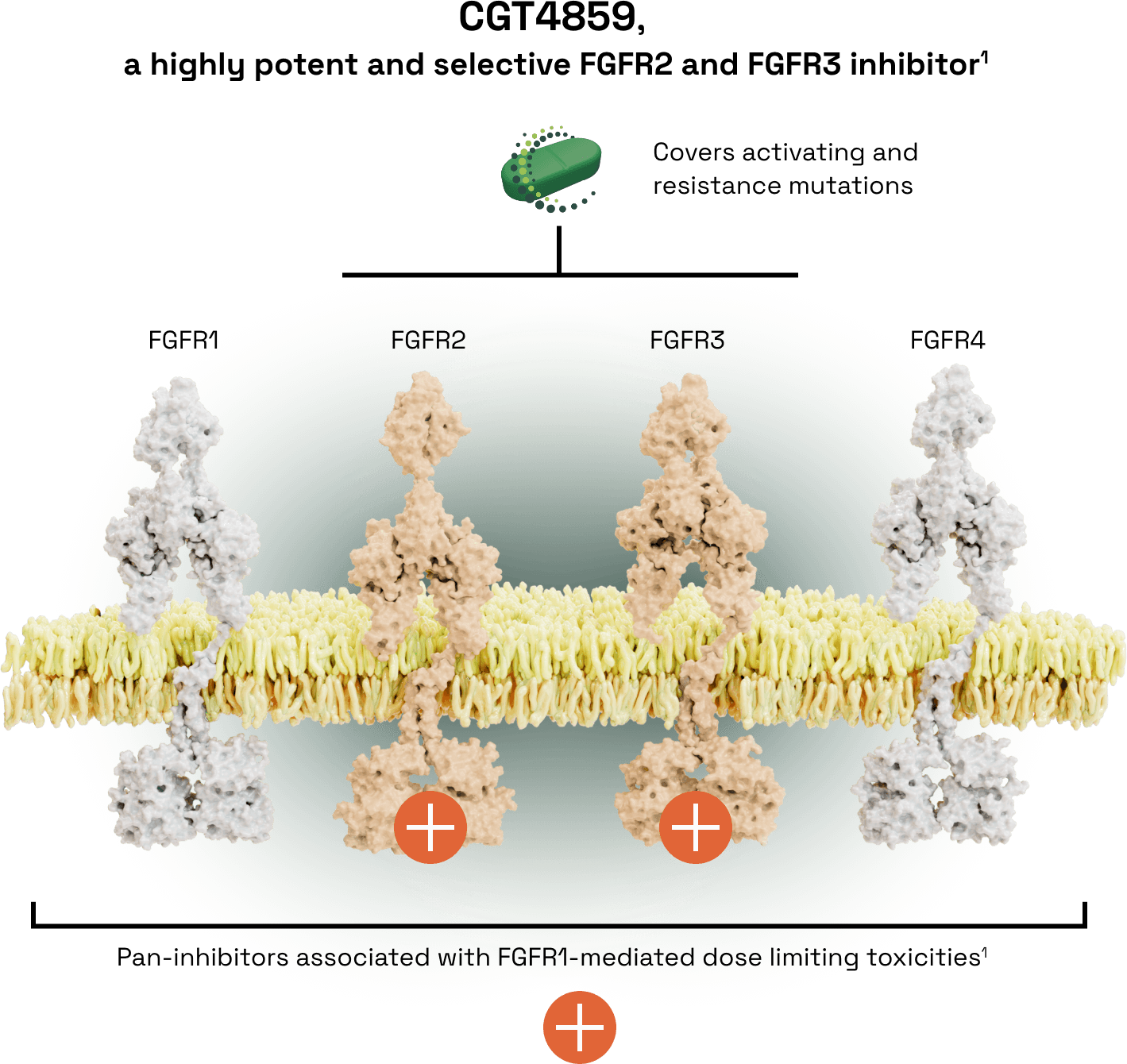
- Approved pan-FGFR inhibitors show on-target, dose-limiting toxicities1
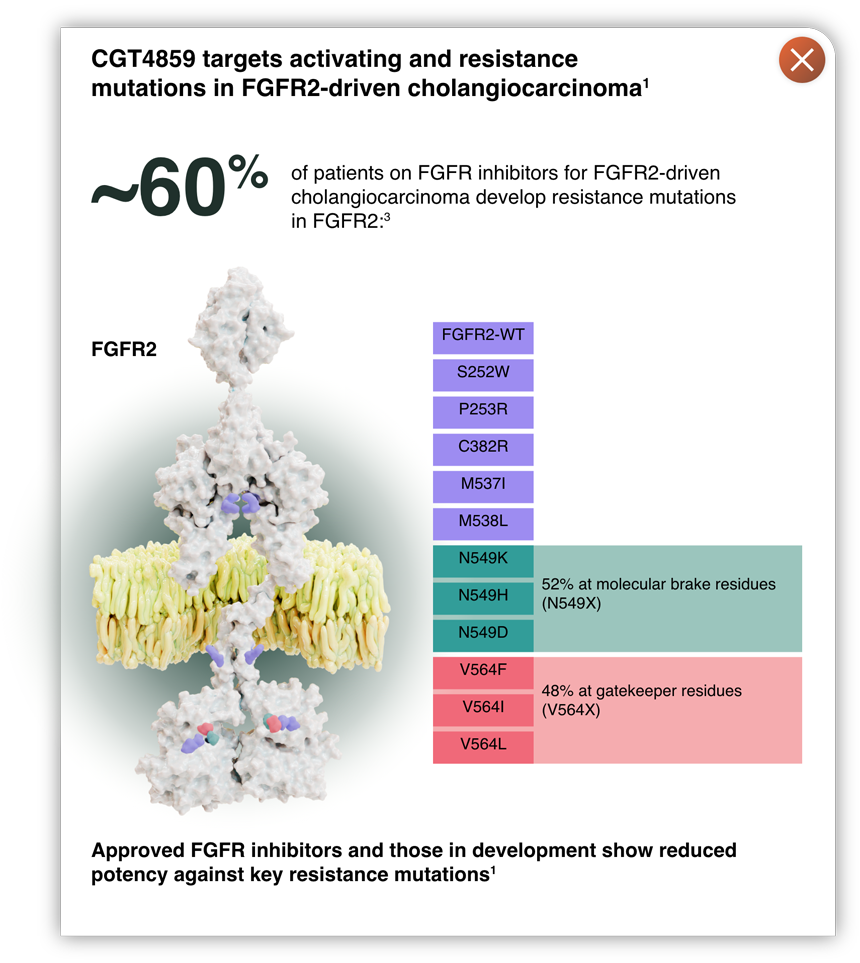
- The FGFR2 gatekeeper and molecular brake residues are common sites of resistance mutations1,3
There is a need for potent, selective FGFR2 inhibitors, with coverage of activating and resistance mutations, avoiding FGFR1-mediated hyperphosphatemia.1
- Avoids FGFR1 inhibition, associated with dose-limiting toxicities such as hyperphosphatemia
- Retains potency against acquired resistance mutations in FGFR2 and FGFR3, including gatekeeper and molecular brake mutations
References
- Fischer J et al. AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics. Poster C161. Boston, 2023.
- Helsten T et al. Clin Cancer Res 2016; 22(1): 259-67.
- Wu Q et al. Clin Cancer Res 2024; 30(1): 198-208.
- Fischer J et al. EORTC-NCI-AACR symposium. Poster PB107. Barcelona, 2022.
- Facchinetti F et al. Cancer Discov 2023; 13(9): 1998-2011.
- GENIE Cohort v17.0 public. The AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine Through An International Consortium, Cancer Discov. 2017; 7(8): 818-31.
RESEARCH Programs

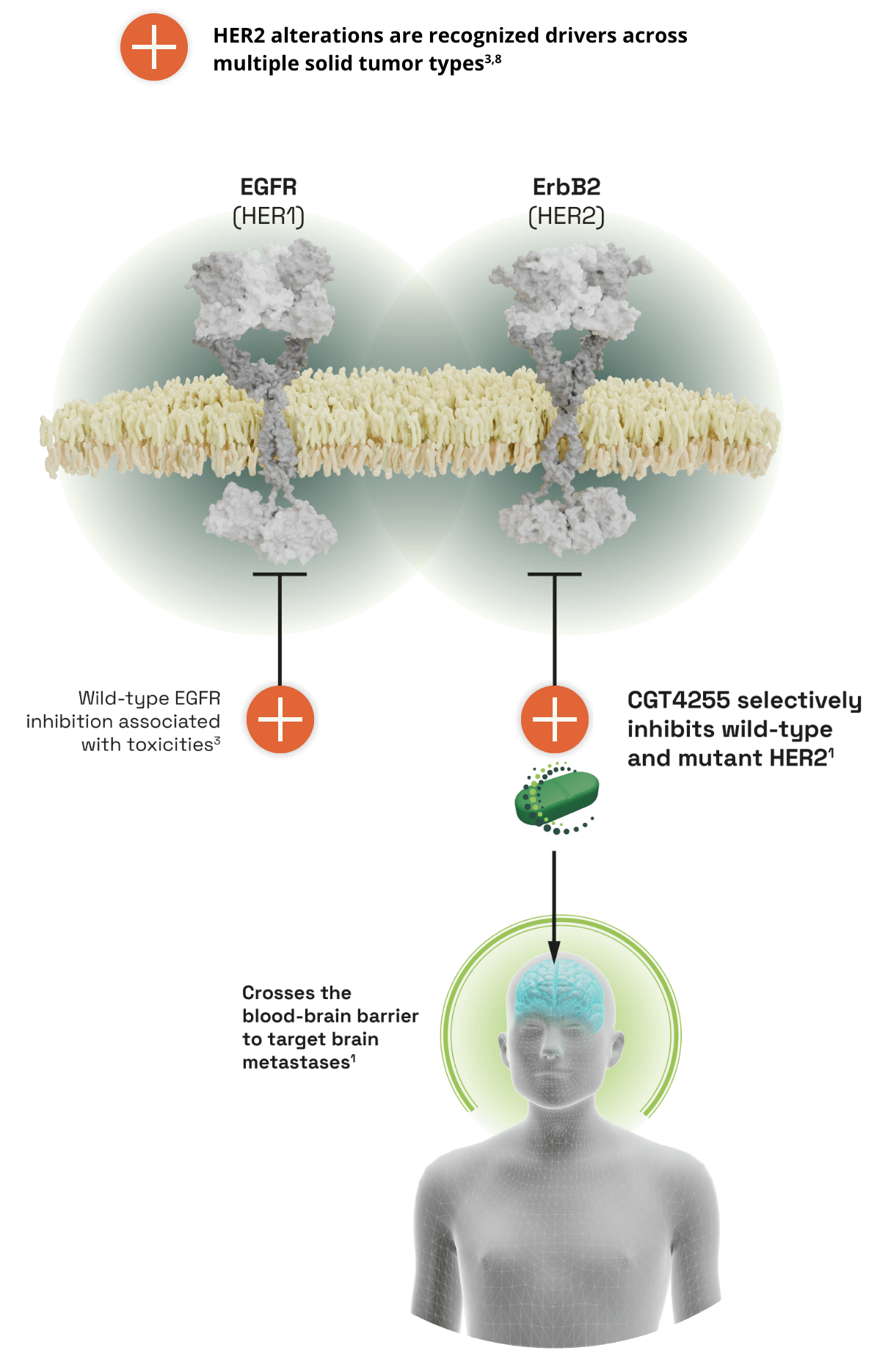
CGT4255, a novel EGFR-sparing ErbB2 inhibitor with best-in-class brain penetration1
Disease Background
- ErbB2 (also known as HER2) belongs to the ErbB tyrosine receptor family which includes EGFR (HER1)2
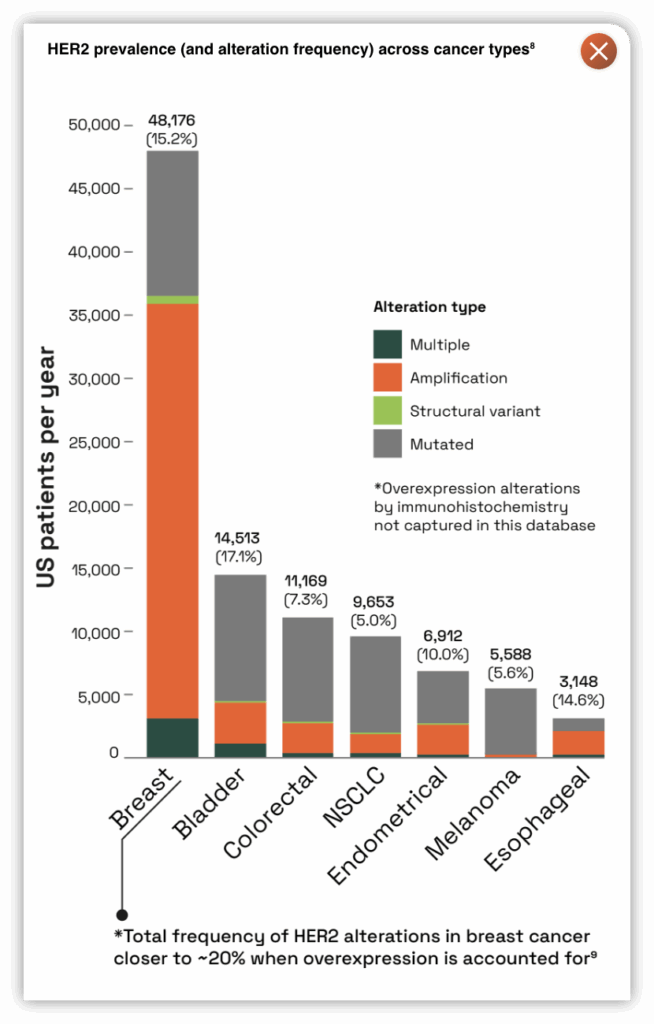
- HER2 gene amplifications and activating mutations are well known oncogenic drivers of tumor growth and are prevalent in breast cancer and non-small cell lung cancer (NSCLC)3.4
- Brain metastases are common in patients with HER2+ metastatic breast cancer and in stage IV/metastatic HER2-mutant NSCLC5,6
THE UNMET NEED IN HER2 TARGETED THERAPY
- Limited brain penetration of current anticancer agents is a drawback in treating HER2+ metastatic disease6
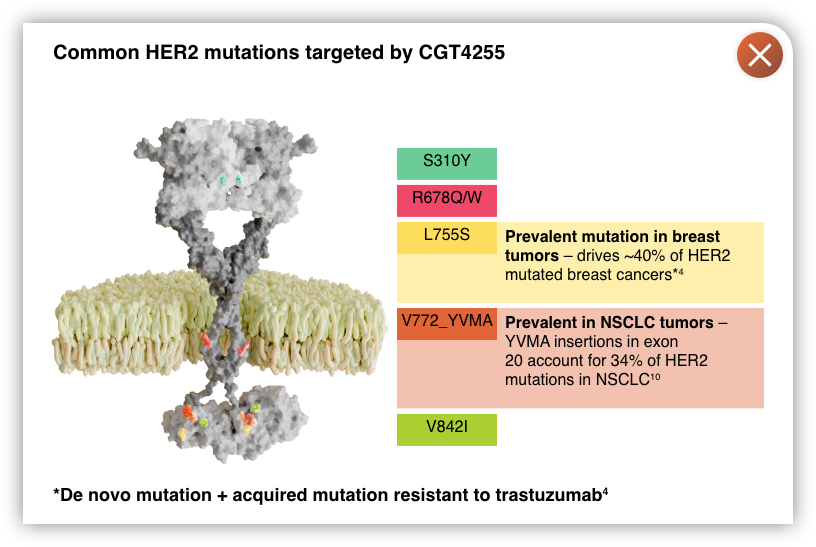
- The breadth of HER2 activating mutations or indels are not covered well by any single approved ErbB2 agent
- Inhibitors with off-target EGFR activity are associated with gastrointestinal and skin toxicities, which can affect quality of life and lead to treatment adjustments3,7
- There is an unmet need for potent HER2 inhibitors that target all prevalent mutations, are selective for HER2 over EGFR, and can penetrate the blood-brain barrier.3
CGT4255, a selective pan-MUTANT HER2 inhibitor that crosses the blood-brain barrier1
- High brain and CSF penetration in preclinical animal models; high predicted brain concentration in humans
- Demonstrates equivalent inhibition of subcutaneous and intracranial tumors
- Well tolerated at efficacious doses in preclinical tumor growth inhibition models
References
- Chicarelli M et al. San Antonio Breast Cancer Symposium. Poster PO3-26-02. San Antonio, 2024.
- Tai W et al. J Control Release 2010; 146(3): 264–75.
- Fulton J et al. AACR Annual Meeting. Poster 1440. Orlando, 2023.
- Nagano M et al. Clin Cancer Res 2018; 24(20): 5112–22.
- Sun H et al. Cancer Med 2023; 12(2): 1007–24.
- Offin M et al. Cancer. 2019; 125(24): 4380–7.
- Shah R and Shah D. Drug Saf 2019; 42(2): 181–98.
- The AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine Through An International Consortium, Cancer Discovery 2017.
- Gutierrez C and Schiff R. Arch Pathol Lab Med. 2011; 135(1): 55–62.
- Ferrari G et al. Cancers (Basel); 16(11): 2018.

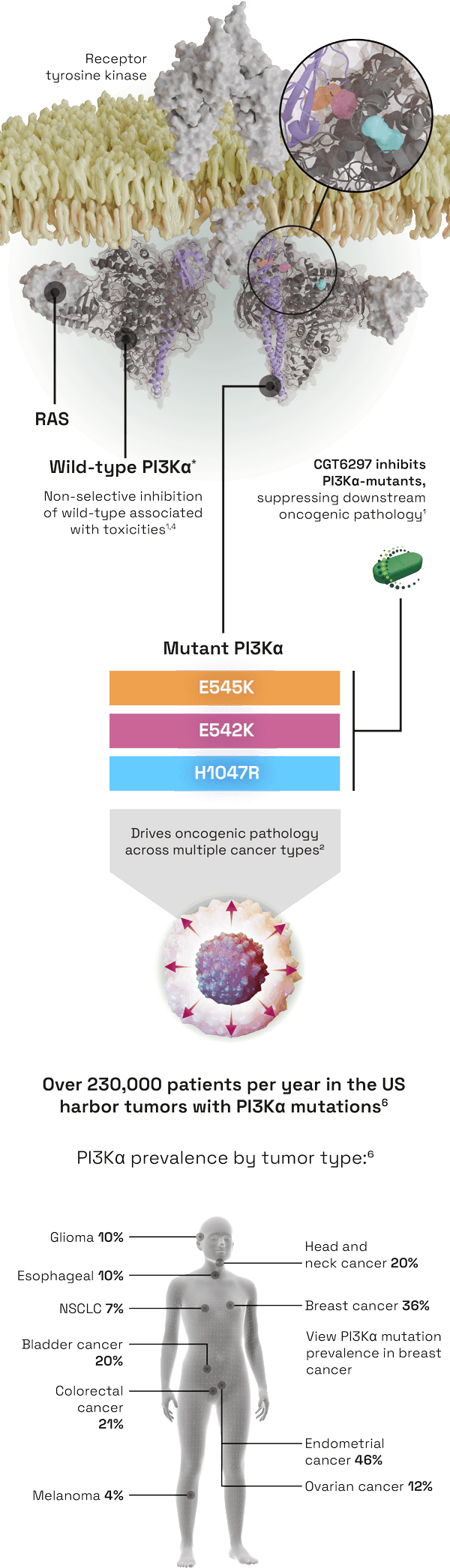
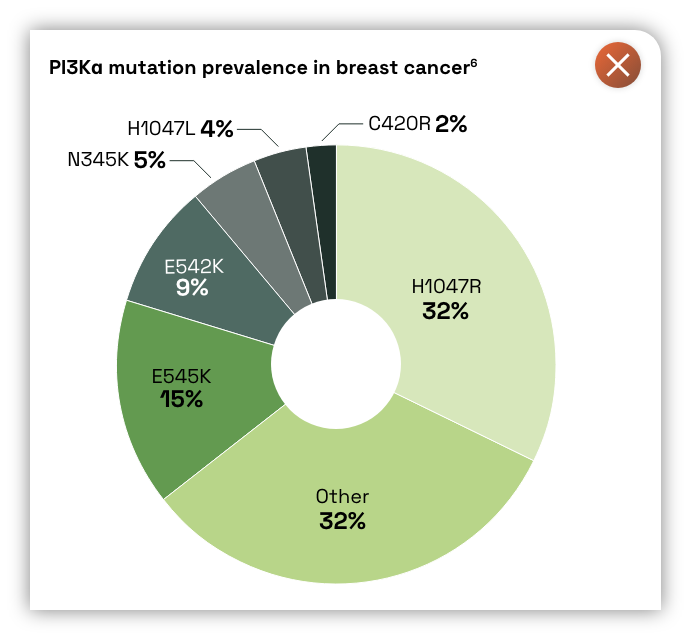
CGT6297 is a potent inhibitor of mutant PI3Kα with selectivity over wild-type PI3Kα1
Disease Background
- PI3Kα is a key component of the PI3K-AKT signaling pathway, which regulates a diverse range of cellular functions: cell growth and survival, angiogenesis, apoptosis and glucose metabolism2
- PI3K is the most frequently mutated kinase, driving 17% of human cancers through sustained activation and dysregulation of the pathway2,3
THE UNMET NEED
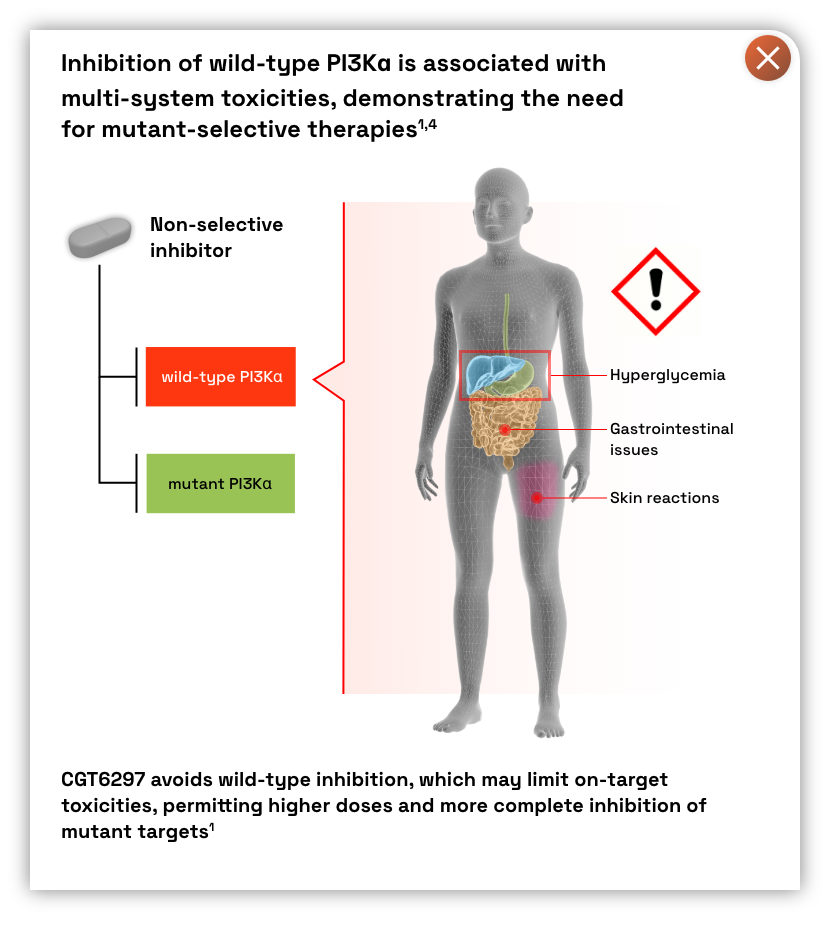
- Approved inhibitors targeting the PI3Kɑ isoform have shown significant anti-cancer benefits; however, on-target inhibition of wild-type PI3Kɑ leads to multi-system toxicities2,4
- There is a need for a mutant-selective inhibitor, providing targeted anti-tumor efficacy while avoiding wild-type associated toxicities.1,4
CGT6297 is a novel PI3Kα mutant inhibitor, with selectivity over wild-type PI3Kα1,5
- Potent inhibition of PI3Kα helical (E542K, E545K) and kinase domain (H1047R) mutations, with demonstrated efficacy in preclinical models at doses that do not cause glucose dysregulation
- Poised to avoid on-target, dose-limiting toxicities associated with wild-type inhibition such as hyperglycemia
References
- Smith A et al. Breast Cancer Symposium. Presentation P4-12-19. San Antonio, 2024.
- Glaviano A et al. Mol Cancer 2023; 22(1): doi.org/10.1186/s12943-023-01827-6
- Wang Y et al. Genes Dis 2024; 12(2): doi: 10.1016/j.gendis.2024.101430.
- Hanker A et al. Cancer Discov 2019; 9(4): 482-91.
- Knox H et al. AACR annual meeting. Poster 5, abstract 3005. Chicago, 2025
- AACR Project GENIE Consortium. Cancer Discov 2017; 7(8): 818-31.
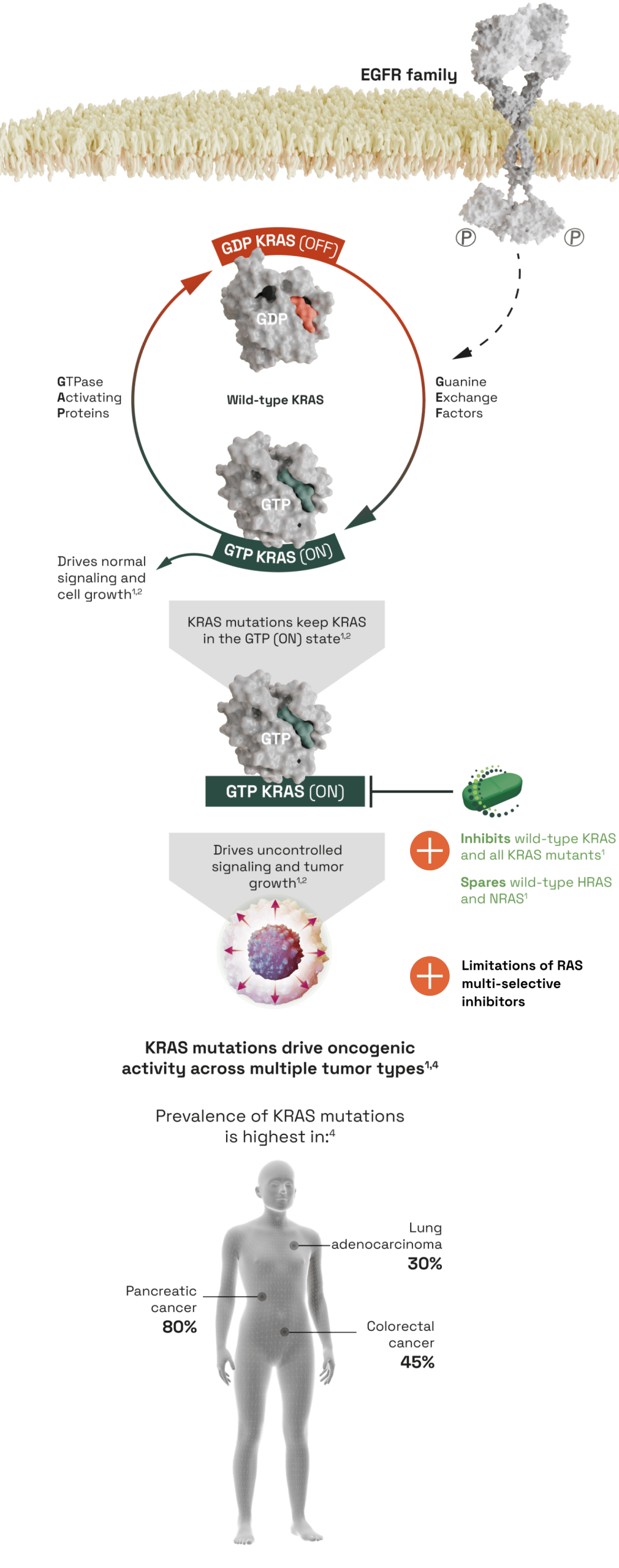
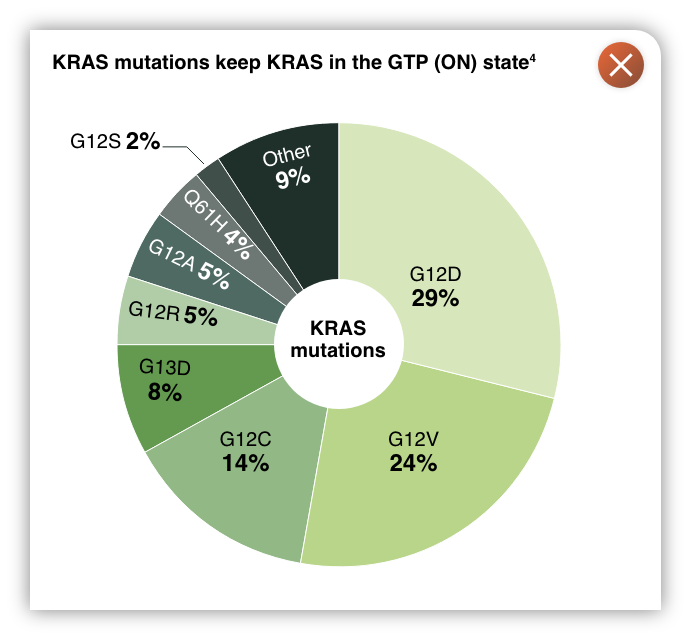
A pan-KRAS inhibitor with potent activity across prevalent KRAS mutations
Disease Background
- KRAS is a small GTPase that acts as a switch that controls proliferation and survival pathways. Activating mutations in KRAS are highly prevalent in cancer, and lead to an accumulation of the GTP KRAS (ON) state, leading to persistent pathogenic activation of downstream signaling2,3
- Activating mutations in KRAS occur in up to 175,000 new cancer patients per year in the U.S., and are particularly prevalent in Colorectal Cancer (CRC), Pancreatic Cancer, and Lung Adenocarcinomas4
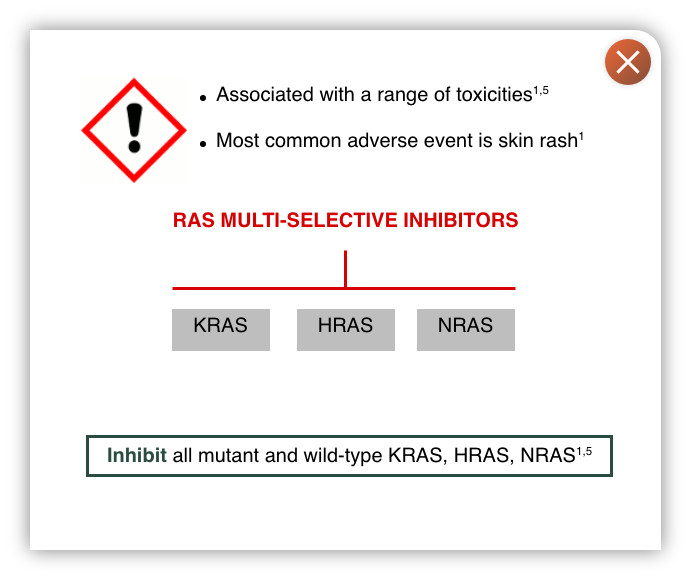
- KRAS is a member of the RAS family of oncogenes that also includes HRAS and NRAS
THE UNMET NEED IN KRAS INHIBITION
- Current approved agents targeting KRAS only address the G12C mutation, which comprises only 14% of KRAS mutations, leaving 150,000 U.S. patients with additional KRAS mutations without an option4
- RAS multi-selective inhibitors have potent activity against KRAS across multiple mutations. However, as they also target HRAS and NRAS they can be associated with a range of toxicities, most commonly skin rash3
- There is a need for potent inhibitors that target multiple KRAS mutations and are selective for KRAS over other RAS isoforms
THE COGENT selective pan-KRAS inhibitor1
- Potently inhibits KRAS across prevalent KRAS mutations, and demonstrates robust tumor regression in preclinical models1
- Is able to bind and inhibit KRAS in both the GTP KRAS (ON) and GDP KRAS (OFF) state1
- Shows >1000x selectivity for KRAS over HRAS and NRAS isoforms, which may confer tolerability advantages over RAS multi-selective inhibitors1
References
- Fischer J et al. EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics. Poster PB108. Barcelona, 2024.
- Moore A et al. Nat Rev Drug Discov. 2020; 19(8): 533–52.
- Drosten M et al. 2014; 33(22): 2857-65.
- The AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine Through An International Consortium, Cancer Discov. 2017; 7(8): 818–31.
- Singhal A et al. Nature Medicine 2024; 30: 969–83.


| wdt_ID | wdt_created_by | wdt_created_at | wdt_last_edited_by | wdt_last_edited_at | INDICATION | DATE | TITLE | JOURNAL/CONFERENCE |
|---|---|---|---|---|---|---|---|---|
| 2 | cmatranga@macdougall.bio | March 2024 08:48 PM | cmatranga@macdougall.bio | March 2024 10:52 PM | Non-Advanced Systemic Mastocytosis | February 2024 | American Academy of Asthma, Allergy & Immunology Annual Meeting | |
| 3 | cmatranga@macdougall.bio | March 2024 08:49 PM | cmatranga@macdougall.bio | March 2024 08:08 PM | Advanced Systemic Mastocytosis | December 2023 | American Society of Hematology Annual Meeting | |
| 4 | cmatranga@macdougall.bio | March 2024 08:50 PM | cmatranga@macdougall.bio | March 2024 08:20 PM | Non-Advanced Systemic Mastocytosis | December 2023 | American Society of Hematology Annual Meeting | |
| 5 | cmatranga@macdougall.bio | March 2024 08:51 PM | cmatranga@macdougall.bio | March 2024 08:22 PM | ErbB2 | December 2023 | San Antonio Breast Cancer Symposium | |
| 6 | cmatranga@macdougall.bio | March 2024 08:52 PM | cmatranga@macdougall.bio | March 2024 08:22 PM | PI3Kα | December 2023 | San Antonio Breast Cancer Symposium | |
| 7 | cmatranga@macdougall.bio | March 2024 08:52 PM | cmatranga@macdougall.bio | March 2024 08:23 PM | Gastrointestinal Stromal Tumors | November 2023 | Connective Tissue Oncology Society Annual Meeting | |
| 8 | cmatranga@macdougall.bio | March 2024 08:53 PM | cmatranga@macdougall.bio | March 2024 08:23 PM | FGFR2 | October 2023 | AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics | |
| 9 | cmatranga@macdougall.bio | March 2024 08:54 PM | cmatranga@macdougall.bio | March 2024 08:25 PM | Gastrointestinal Stromal Tumors | June 2023 | American Society of Clinical Oncology Annual Meeting | |
| 10 | cmatranga@macdougall.bio | March 2024 08:55 PM | cmatranga@macdougall.bio | March 2024 08:25 PM | ErbB2 | April 2023 | American Association for Cancer Research Annual Meeting | |
| 11 | cmatranga@macdougall.bio | March 2024 08:55 PM | cmatranga@macdougall.bio | March 2024 08:26 PM | FGFR2 | April 2023 | American Association for Cancer Research Annual Meeting |